


Ernst Jan Eggers1,2, Ate van der Burgt1, Sjaak AW van Heusden2, Michiel E. de Vries@ 1, Richard GF Visser©2, Christian WB Bachem©2и y Pim Lindhout1
La ganancia genética en la papa se ve obstaculizada por el genoma tetraploide heterocigótico de la papa cultivada. Convertir la papa en un cultivo híbrido F1 basado en una línea endogámica diploide proporciona una ruta prometedora hacia una mayor ganancia genética. La introducción de un gen inhibidor del locus S dominante (Sli) en el germoplasma de papa diploide permite la generación eficiente de semillas autofecundadas y, por lo tanto, el desarrollo de líneas puras de papa. Poco se sabe sobre la estructura y función del locus de Sli. Aquí describimos el mapeo de Sli a un intervalo de 12.6 kb en el cromosoma 12 utilizando un enfoque de pantalla recombinante. Uno de los dos genes candidatos presentes en este intervalo muestra una secuencia única que está presente exclusivamente en líneas autocompatibles. Describimos un vector de expresión que convierte auto-incompatibles genotipos en autocompatible y un vector CRISPR-Cas9 que convierte los genotipos SC en SI. El gen Sli codifica una proteína F-box que se expresa específicamente en el polen de plantas autocompatibles. Una inserción de 533 pb en el promotor de ese gen conduce a una mutación de ganancia de función, que supera el rechazo del propio polen.
La papa es el cultivo alimentario no cereal más importante del mundo. Sin embargo, mientras que otros cultivos alimentarios como el maíz, el arroz y el trigo han mostrado un aumento del rendimiento genético del 1 % anual1, la ganancia genética en papa ha sido mínima2. Actualmente, la mayoría de los cultivares de papa cultivados comercialmente se derivan de cruces entre padres heterocigotos autotetraploides. En este sistema de mejoramiento, se generan y analizan cientos de miles de plántulas en cada generación de mejoramiento para identificar aquellos individuos raros que tienen características aceptables para numerosos rasgos que se segregan en la progenie. Como hay unos cincuenta rasgos que son relevantes para el valor de un cultivo de patata comercial, la posibilidad de combinar los mejores alelos que controlan estos rasgos mediante el cultivo convencional de patata es insignificante. Además, la introducción específica de nuevos rasgos en cultivares de élite mientras se mantiene la integridad genética a través de esquemas de retrocruzamiento es imposible sin líneas parentales homocigóticas. Para superar estos problemas, varios grupos han iniciado programas de mejoramiento de papas diploides basados en líneas endogámicas.2–5. En estos programas, las ganancias genéticas se logran a través de mejoras incrementales de las líneas parentales seleccionando continuamente los alelos perjudiciales durante la consanguinidad y apilando los alelos beneficiosos en las líneas endogámicas a través de esquemas de retrocruzamiento.6. Las líneas endogámicas parentales luego se cruzan para producir descendencia híbrida heterótica F1.
En la mayoría de los genotipos diploides de papa, la consanguinidad está severamente limitada por un sistema de autoincompatibilidad gametofítica (GSI) que está controlado por el locus S multialélico. Este locus S codifica las S-RNasas expresadas en el estilo que inhiben el crecimiento del tubo polínico propio en el estilo, evitando la autofecundación.7. Durante la polinización cruzada, las proteínas S-locus F-box (SLF) expresadas en el polen reconocen las S-RNasas y las dirigen a la vía de degradación proteasomal, lo que permite el crecimiento del tubo polínico hacia los ovarios, donde puede tener lugar la fertilización.8. Cada alelo S codifica una S-RNasa y múltiples SLF con diferentes especificidades, que juntas pueden reconocer todas las S-RNasas excepto la S-RNasa que está presente en el mismo alelo.9.
Aunque la mayoría de las líneas de papas diploides son autoincompatibles (SI), existen líneas de papas diploides autocompatibles y se pueden usar para introducir la autocompatibilidad en los programas de mejoramiento de papas diploides.10–12. Hosaka y Hanneman mapearon un inhibidor del locus S dominante (Sli) gen de un Solanum chacoense accesión en el extremo distal del cromosoma 12 y lo usó para generar líneas puras de papa13'14. Con base en sus resultados, Hosaka y Hanneman sugirieron que SLI es un gen expresado en polen con acción esporofítica y esa homocigosidad para SLI es letal ya que homocigoto SliSli Los genotipos estaban ausentes en la población F8 de S. chacoense Usamos uno de estos S. chacoense Líneas puras derivadas de (DS) para introducir autocompatibilidad en S. Tuberosum antecedentes. Aquí, describimos la identificación del gen causal de la autocompatibilidad para obtener más información sobre la biología de la autocompatibilidad en la papa diploide.
Resultados y discusión
En una población F2 derivada de un cruce entre el SLI donante (designado DS) y un diploide S. Tuberosum (D2) observamos un QTL de efecto modesto para el conjunto de bayas propias en el cromosoma 2, pero una selección recombinante posterior no tuvo éxito. Nos dimos cuenta de que múltiples poblaciones F2 mostraban una asimetría extrema alrededor del brazo largo del cromosoma 12, con una ausencia total de homocigosidad para el haplotipo no DS.
Sobre la base de esta asimetría y el mapeo de SLI en el cromosoma 12, planteamos la hipótesis de que SLI se expresa gametofíticamente, lo que significa que en una autopolinización de una planta heterocigota para SLI (Sli/sli), sólo el polen que contiene el dominante SLI el alelo puede participar en la autofecundación. Para probar esta hipótesis, cruzamos una línea vigorosa y altamente autofértil (16HP1-66) con una línea vigorosa autoincompatible (D16), secuenciamos el genoma completo de ambos y analizamos la población F1 resultante (17SC11, n = 251, figura. 1a, b). Como esta población F1 es altamente polimórfica para muchos loci, observamos una amplia gama de fenotipos, incluidos los que están relacionados con la fertilidad. Por lo tanto, implementamos un protocolo de fenotipado muy estricto y riguroso que incluye el conjunto de bayas y semillas de polinizaciones cruzadas y autofecundadas, así como la visualización del crecimiento del tubo polínico en los estilos para evitar problemas de esterilidad que confundan el fenotipo de compatibilidad. Las plantas que producen más de una baya propia se consideran SC, mientras que las plantas que no producen bayas propias después de al menos 10 autopolinizaciones, muestran una detención del crecimiento del tubo polínico propio en el estilo y establecen bayas cruzadas después de la polinización con semillas a granel. el polen se considera SI. Como resultado, una parte importante de la población quedó excluida de los análisis genéticos ya que no se cumplían los requisitos para evaluar sin ambigüedades el fenotipo de compatibilidad. Aún así, se pudo evaluar el estado de compatibilidad de la mayoría de la progenie de la población 17SC11 y se demostró que segrega para la autocompatibilidad (Datos complementarios 1). Dado que la autocompatibilidad se originó en 16HP1-66, utilizamos las secuencias del genoma completo de este genotipo para diseñar marcadores KASP dirigidos a SNP en los cromosomas 2 y 12 que son heterocigotos en 16HP1-66 pero homocigotos en D16, lo que permite el mapeo de SLI en la meiosis materna. Construimos un mapa genético, realizamos un análisis de QTL y encontramos un QTL altamente significativo (LOD = 75.72) en el brazo largo del cromosoma 12 (Fig. 1b), confirmando los resultados de Hosaka y Hanneman.
Para confirmar el QTL en un fondo genético diferente, cruzamos otro genotipo altamente autofértil derivado del programa de mejoramiento de Solynta con el genotipo SI D14 y analizamos la población F1 resultante (17SC25, Datos complementarios 1). Entre 32 individuos de la población 17SC25, no encontramos individuos SI. Para generar una población segregante, seleccionamos el genotipo más fértil y lo cruzamos con dos genotipos SI que identificamos en la población 17SC11, lo que resultó en las poblaciones 18SC11 y 18SC12 (Datos complementarios 1 y Figura complementaria 1). Como era de esperar, el análisis de las poblaciones 18SC11 y 18SC12 mostró que ambas poblaciones se segregan por autocompatibilidad. Enviamos a la madre (17SC25-8) para la secuenciación del genoma completo y usamos estos datos para diseñar nuevos marcadores KASP usando el mismo enfoque que se usó para la población 17SC11, pero esta vez apuntando solo al cromosoma 12. El análisis posterior de QTL confirmó el QTL que encontramos en la población. 17SC11 con valores LOD de 33.14 y 120.94 en las poblaciones 18SC11 y 18SC12, respectivamente (Fig. 2 complementaria).
Para determinar si SLI de hecho se expresa gametofíticamente, analizamos una población F2 (19SC1, n = 160) derivado de un individuo 17SC11 fértil y vigoroso. La floración y la fertilidad en esta población se redujeron en comparación con la F1. En el análisis fenotípico, de 160 plantas, 81 plantas fueron autocompatibles, 78 se clasificaron como no determinadas (ND) debido a la mala floración o la mala fertilidad y una planta se calificó como autoincompatible (Datos complementarios 1). Diseñamos marcadores KASP dirigidos a SNP en el cromosoma 12 que son homocigotos para alelos alternativos en los padres 16HP1-66 y D16. A lo largo de la totalidad de las proporciones de segregación del cromosoma 12 se desvían significativamente de la segregación 1:2:1 esperada. Además, alrededor del QTL de autocompatibilidad, no hay loci homocigotos para el haplotipo del padre D16 (Fig. 3 complementaria), mostrando en cambio una segregación 1: 1 para heterocigotos D16 / 16HP1-66: homocigotos 16HP1-66, lo que sugiere que la eliminación de falta de polen SLI provoca la distorsión de la segregación. Esto apoya la hipótesis de que sólo el polen que lleva la dominante SLI el alelo participa en la autofecundación. Además, aparte de un individuo, el fenotipado fue concluyente para el contraste entre SI y SC, mostrando que el protocolo de fenotipado utilizado es robusto y casi libre de errores.
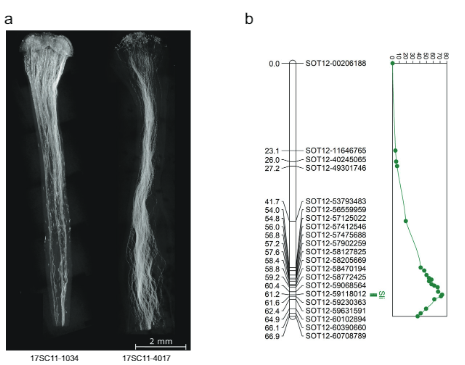
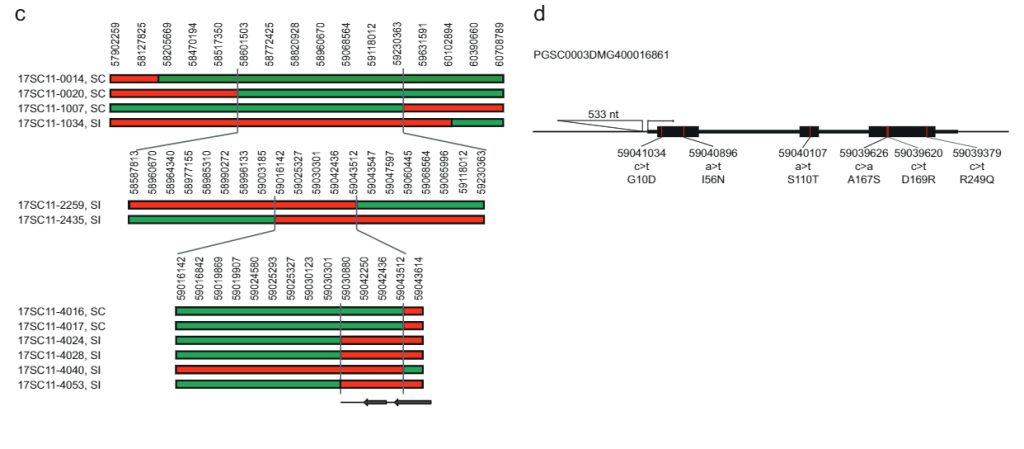
Mientras que los 628 KB SLI El intervalo en el cromosoma 12 que lleva el alelo Sli de la población 17SC11 se redujo a un intervalo superpuesto más pequeño de 169 KB en la población 18SC12, estos intervalos aún eran demasiado grandes para identificar el gen Sli. Por lo tanto, nuestro objetivo era reducir el intervalo que contenía S/i a través de un enfoque de detección recombinante. Identificar plantas con una recombinación en el SLI intervalo, genotipamos 1374 plántulas 17SC11 con dos marcadores KASP en el borde proximal y dos en el borde distal. Identificamos 81 plántulas con recombinación entre los dos marcadores más externos y las seleccionamos para un mapeo más fino. Para obtener fenotipos inequívocos, propagamos vegetativamente estos genotipos y realizamos el fenotipado en al menos dos clones por genotipo. Genotipamos los 81 recombinantes con más marcadores en el intervalo e identificamos dos recombinantes informativos que redujeron el intervalo a 27.37 KB que contenían cinco genes anotados (Datos complementarios 1).
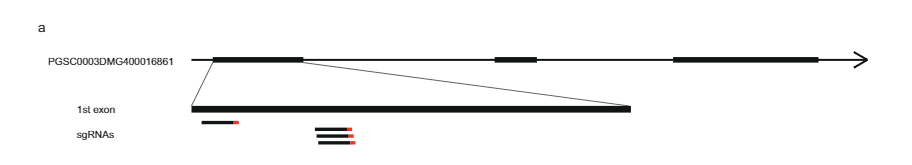
Para reducir aún más el intervalo, seleccionamos otras 10165 plántulas de la misma población con cuatro marcadores alrededor de este intervalo de 27.37 KB e identificamos 12 recombinantes. Estos se genotipificaron adicionalmente con 14 marcadores más en este intervalo e identificamos seis recombinantes informativos que mostraron fenotipos de compatibilidad clara. Dos recombinantes con fenotipo SC y uno con fenotipo SI confirmaron el borde distal del intervalo de 27.37 KB, mientras que tres recombinantes con fenotipo SI definieron un nuevo borde proximal que reduce el intervalo a solo 12.6 kb que contienen dos genes, PGSC0003DMG400016861 y PGSC0003DMG400016860 ( Higo. 1do).
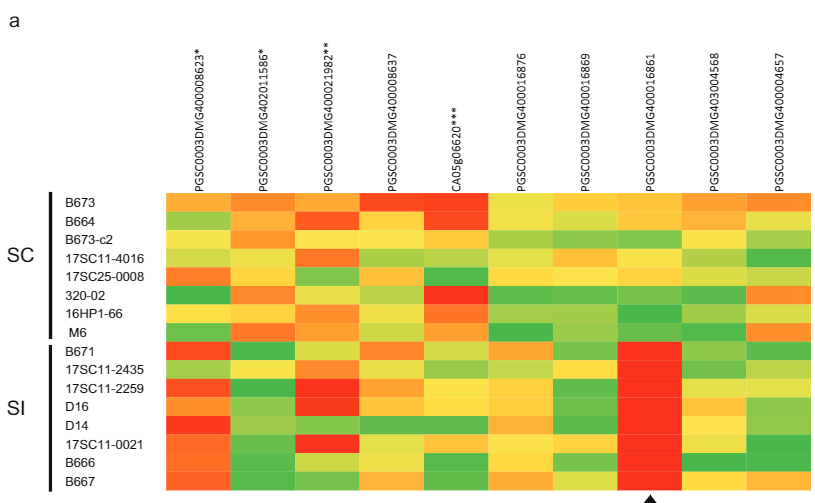
Para identificar el gen candidato que es responsable del fenotipo autocompatible, analizamos la variación de la secuencia de estos dos genes en varias líneas de papa diploide secuenciadas del genoma completo (Datos complementarios 2). Al comparar esta variación de secuencia con los fenotipos SC / SI de estas líneas, identificamos todos los SNP e INDELS específicos de SC (Datos complementarios 2). A continuación, identificamos manualmente todos los SNP no sinónimos y determinamos si las sustituciones de aminoácidos son comunes o únicas para proteínas similares en las solanáceas. El gen candidato PGSC0003DMG400016861 muestra seis sustituciones de aminoácidos específicas de SC y, en particular, una inserción de 533 pb ubicada a -108 pb desde el codón de inicio, lo que sugiere que el alelo SC tiene una expresión alterada en comparación con el alelo SI. Con base en estos estudios genéticos, planteamos la hipótesis de que PGSC0003DMG400016861 es el SLI .
Para verificar la hipótesis de que Sli se expresa en el polen, germinamos polen de genotipos de papa 10 SI y 10 SC in vitro y extrajimos el ARN para la secuenciación del ARN. De los dos genes candidatos restantes, solo se expresó el gen candidato PGSC0003DMG400016861, pero exclusivamente en polen de genotipos SC (Fig. 2a). Además, en plantas heterocigóticas para el gen candidato putativo Sli, sólo se expresó el alelo Sli. Curiosamente, otros genes expresados en polen ubicados cerca del locus Sli en el cromosoma 12 mostraron niveles de expresión similares en plantas SC y SI (Fig. 2a). Por lo tanto, concluimos que solo el gen PGSC0003DMG400016861 se expresa específicamente en los tubos polínicos de plantas SC.
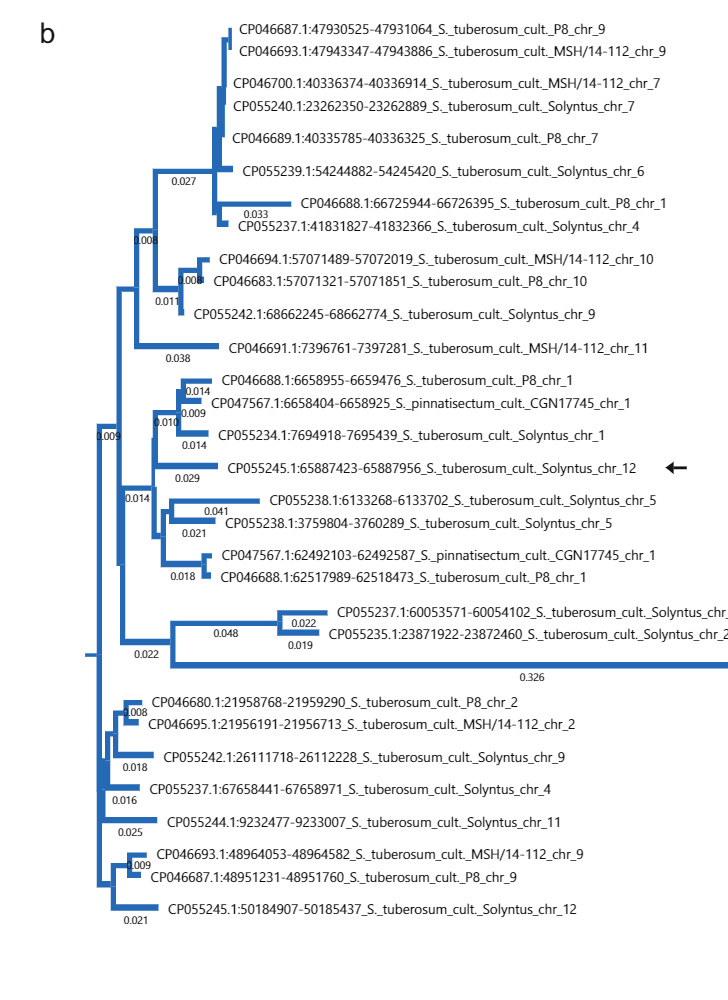
Para investigar el origen de la inserción de 533 pb, realizamos una búsqueda BLAST de la secuencia de 533 pb en NCBI. Curiosamente, las secuencias notablemente similares a la inserción específica de Sli son comunes en S. Tuberosum accesiones (Fig. 2b). Además, la inserción de 533 pb tiene una homología con una secuencia en S. pennellii. Usando la secuencia de S. pennellii como una consulta BLAST encontramos secuencias similares en S. lycopersicum. Análisis filogenético de las secuencias en S. tuberosum, S.pennellii y S. lycopersicum agrupa el S.pennellii secuencia junto con el S. lycopersicum y un experto S. tuberosum secuencia, lo que sugiere que comparten un origen común (Fig. 4a complementaria). Presumimos que la inserción se deriva de un elemento transponible (TE). Generamos un gráfico de puntos a partir de la secuencia de 533 pb y observamos que la secuencia contenía repeticiones invertidas en miniatura (Fig. 4b complementaria). Enviamos la inserción de 533 pb a BLAST contra la base de datos MITE de la planta, lo que resultó en múltiples aciertos de la familia MITE DTA_Sot42 en S. tuberosum15, indicando que la inserción de 533 pb en el promotor de SLI de hecho, se origina en un TE (Fig. 4c complementaria).
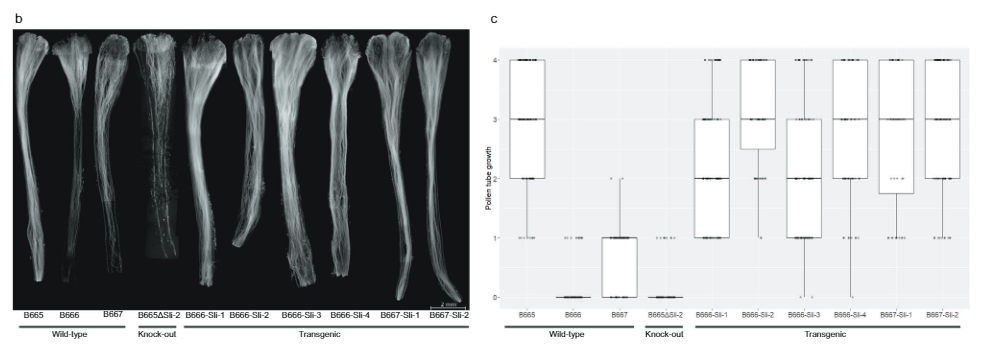
Para confirmar aún más que PGSC0003DMG400016861 es de hecho SLI, diseñamos un constructo de expresión que contiene los exones del alelo SC de SLI entre su promotor nativo y terminador (Fig. 3a) en el vector pBINPLUS (pBINPLUS-Sli). Usamos esta construcción para transformar dos genotipos SI a partir del mapeo de la población 18SC12. Fenotipamos de dos a seis clones de cada uno de los cinco transgénicos independientes derivados del genotipo SI B666 y tres transgénicos derivados del genotipo SI B667.
Los clones de seis transgénicos independientes establecen fácilmente bayas en la autopolinización (Datos complementarios 3). Además, la microscopía de fluorescencia mostró que el polen de SLI las plantas transgénicas se vuelven más profundas en estilos propios que los controles no transformados (Fig. 3antes de Cristo). Calificamos el crecimiento del tubo polínico en 442 autopolinizaciones en SLI transgénicos y 179 autopolinizaciones en controles no transformados en una escala de 0-4. La mayoría de los tubos polínicos llegaron a los ovarios en la mayoría SLI transgénicos, en comparación con solo una fracción muy pequeña de los controles, lo que indica que PGSC0003DMG400016861 es el SLI .
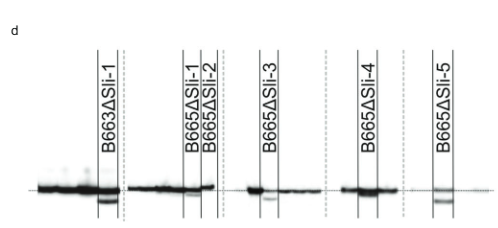
A continuación, diseñamos una construcción CRISPR-Cas9 que codifica cuatro gRNA dirigidos al primer exón de PGSC0003DMG400016861 (pAGM: CRISPRASli, Fig. 3a). Transformamos dos genotipos SC (B665 y B663) con esta construcción y obtuvimos 149 regenerantes transformados. Luego analizamos el exón objetivo usando PAGE para identificar los INDEL inducidos por CRISPR-Cas9. Desafortunadamente, el vector pAGM:CRISPRASli tuvo una baja eficiencia, solo seis de los 149 regenerantes mostraron INDELs en SLI (Higo. 3d). Cinco de estas líneas CRISPR-Cas9 son heterocigotas para los INDEL, pero una línea, B665ASli-2, es homocigota para un INDEL pequeño. Mientras que B665 no transformado establece fácilmente bayas propias y muestra un buen crecimiento del tubo polínico propio a través de 105 estilos observados, B665ASli-2 no establece bayas en la autopolinización y su polen no puede crecer a través de 78 estilos observados (Fig. 3b, c y Datos complementarios 3), proporcionando más evidencia de que PGSC0003DMG400016861 es de hecho el SLI .
En los sistemas de autoincompatibilidad gametofítica basados en S-RNasa, la autofecundación se evita mediante S-RNasas expresadas en pistilo que entran en los tubos polínicos y ejercen efectos citotóxicos en el polen propio o en cualquier otro polen que carezca de una caja S-Locus F correspondiente (SLF ) proteína. La fertilización cruzada está habilitada por las proteínas SLF expresadas en el polen que pueden reconocer y desintoxicar las S-RNasas no propias. Cada alelo S codifica múltiples SLF, cada uno de los cuales puede reconocer una S-RNasa diferente, y juntos pueden reconocer la mayoría de las S-RNasas presentes en la patata, excepto la S-RNasa codificada en el mismo alelo S. Sli codifica una proteína de caja F PP2-B10, que consta de un dominio de caja F unido a un dominio de lectina. Se sabe que los dominios de lectina interactúan con los carbohidratos y podrían interactuar con las proteínas glicosiladas.16. Además, se ha demostrado que las S-RNasas están glicosiladas17. Presumimos que la inserción de 533 pb en el promotor del alelo SC de Sli permite la expresión en el polen, donde Sli puede unirse y desintoxicar las S-RNasas propias, lo que lleva a la pérdida de la detención del crecimiento del tubo polínico y, por lo tanto, a la autocompatibilidad. Una investigación detallada sobre la actividad de MITE en papas realizada por Laimbeer encontró que el 2% de las inserciones de MITE cerca de las regiones génicas estaban asociadas con alteraciones en la expresión génica.
Además, de 1935 inserciones de hAT probadas cerca de regiones génicas, 13 dieron como resultado una regulación positiva del gen asociado, lo que indica que la expresión alterada específica del polen de SLI de hecho podría ser causado por la inserción de 533 pb en su promotor18. Sin embargo, se requiere más investigación para determinar la validez de esta hipótesis.
Previamente, Clot et al utilizaron un enfoque de mapeo de K-mer segregante a granel para identificar un intervalo de 333 kb en el cromosoma 12 en el que se debe ubicar Sli19. Aquí, asignamos el locus de Sli a la misma región del cromosoma 12 en una población F1 y usamos un cribado recombinante para reducir el intervalo a 12.6 KB que contenían 2 genes. El análisis de expresión reveló que el alelo SC de uno de estos genes se expresa específicamente en el polen de los genotipos SC. Finalmente, utilizando la expresión transgénica y la desactivación inducida por CRISPR-Cas9, mostramos de manera concluyente que PGSC0003DMG400016861 es Sli. Si bien el estudio de Laimbeer mostró que los MITE pueden regular al alza los genes próximos de una manera específica del tejido, se requiere más investigación para demostrar que la presencia del MITE en el promotor Sli es la causa de su expresión específica del polen.
En los materiales investigados en este estudio (Datos complementarios 4), no pudimos confirmar informes anteriores de letalidad asociada con la homocigosidad para Sli, ya que encontramos plantas F2 viables homocigóticas para Sli que pudieron establecer bayas (Datos complementarios 1)12>20. Además, las secuencias del genoma de la línea endogámica Solyntus, así como la línea endogámica M6, muestran que ambas líneas son homocigotas para Sli, lo que indica que la homocigosis para Sli en sí misma no es letal, aunque todavía es posible que una letal. el alelo ligado genéticamente a Sli en un ancestro se ha eliminado mediante recombinación en estos genotipos21'22. Sin embargo, a partir de los datos generados en este estudio, no podemos excluir la posibilidad de que la distorsión de segregación observada en la población F2 sea causada por un alelo letal ligado en fase al alelo SI de Sli. Hasta el momento, no está claro si Sli en sí mismo puede reconocer y desintoxicar directamente las S-RNasas. Además, no está claro si Sli da como resultado la autocompatibilidad en todos los genotipos del locus S. Es posible que Sli no pueda inhibir la función de algunos alelos S. Se necesita más investigación para resolver esta pregunta.
Si bien la identificación de Sli permite aún más la reproducción híbrida basada en líneas endogámicas utilizando papa diploide, quedan otros obstáculos. En primer lugar, lo más significativo es que la papa diploide sufre de depresión endogámica, lo que conduce a una reducción del vigor y la fertilidad tras la consanguinidad. La purga de alelos nocivos mediante la autofertilización continua de líneas de papa diploides es un método eficaz para reducir la depresión por endogamia y ya ha resultado en la generación de líneas endogámicas de papa comparativamente vigorosas y fértiles.2'23–25. En segundo lugar, el donante de Sli utilizado en este estudio, DS, se deriva de una S. chacoense adhesión, lo que podría generar problemas con el arrastre de ligamiento de alelos nocivos de S. chacoense En el programa de mejoramiento diploide de Solynta, no vemos problemas aparentes debido a alelos nocivos provenientes de S. chacoense. Además, un estudio reciente sobre autocompatibilidad reveló que los k-mers específicos de SC ya están presentes en varios cultivares tetraploides, lo que proporciona una ruta para eludir este posible arrastre de enlace por completo mediante el uso de dihaploides generados a partir de estos cultivares como donantes de Sli.19.
Métodos
Materiales vegetales. Todos los materiales vegetales utilizados se enumeran en Datos complementarios 4
Condiciones de invernadero. Todas las plantas se cultivaron en invernaderos que se calentaban cuando la temperatura descendía por debajo de los 14 °C y se enfriaban abriendo las ventanas cuando la temperatura subía por encima de los 19 °C. La iluminación artificial complementó la luz natural cuando la intensidad de la luz cayó por debajo de 85 W/M2. Las plantas se cultivaron en una mezcla de sustrato de patata especial de Lentse Potgrond (Lentse Potgrond BV, Katwijk, Países Bajos). La mezcla de sustrato utilizada se compone de una mezcla de turba para una absorción de agua equilibrada, fertilizante básico de liberación lenta y cal para garantizar el nivel de pH requerido. La mezcla de sustrato se fertilizó utilizando una solución de nitrógeno: fósforo: potasio 20:20:20 con una conductividad eléctrica (CE) de 1.5.
Evaluación de la autocompatibilidad. Las flores y los capullos se contaron una vez por semana y el vigor se puntuó una vez por mes en una escala del 1 al 9, siendo 1 una planta extremadamente poco vigorosa y 9 una planta extremadamente vigorosa. El polen de múltiples flores de una planta se recogió en un tubo Eppendorf y se usó inmediatamente para la autopolinización en las mismas flores con un máximo de 10 flores por planta por semana. Las plantas que produjeron más de dos bayas propias que contenían al menos 35 semillas por baya propia se clasificaron como autocompatibles. Para determinar la fertilidad femenina, las plantas se polinizaron con polen a granel de al menos tres genotipos no relacionados. Las plantas que no produjeron bayas propias después de al menos 10 autopolinizaciones, pero que produjeron al menos una baya a granel y mostraron polen fértil en el análisis microscópico de estilos autopolinizados se clasificaron como autoincompatibles. Para 40 genotipos de las poblaciones de mapeo en las que los datos de conjuntos de semillas y bayas no fueron concluyentes (17SC11: n = 14, 18SC11: n = 7, 18SC12: n = 19), la clasificación fenotípica se basó en el crecimiento del tubo polínico propio a través de los estilos (anotado en Datos complementarios 1).
Imágenes de estilo. Para visualizar el crecimiento del tubo polínico, los estilos polinizados se retiraron 24-48 h después de la polinización y luego se fijaron en etanol:ácido acético 3:1 durante al menos 24 h. Luego, los estilos se maceraron en NaOH 8M durante 10 min a 65 °C y se enjuagaron dos veces con agua desionizada. Los estilos se colocaron en portaobjetos de microscopía y se tiñeron durante 2 a 5 minutos con azul de anilina al 0.1 % (Carl Roth GmbH) en 0.1 MK.4P2O7 (pH = 7), luego aplastado en glicerol usando un cubreobjetos y observado usando un microscopio de fluorescencia Zeiss Axiolab usando un juego de filtros 01 (BP 365/12, FT 395 y LP 397). Todos los estilos se observaron y puntuaron usando dos parámetros: (1) penetración más profunda en el estilo, expresada en porcentaje de penetración máxima, (2) % de tubos polínicos que alcanzaron la penetración más profunda. Luego convertimos estos porcentajes a una escala de 0 a 4, donde los estilos en los que ningún tubo polínico llegaba al ovario obtuvieron una puntuación de 0, los estilos en los que entre el 0 y el 25 % de los tubos polínicos obtuvieron una puntuación de 1, los estilos en los que entre 25 y el 50% de los tubos polínicos llegaron al ovario obtuvieron una puntuación de 2, los estilos en los que entre el 50 y el 75% de los tubos polínicos llegaron al ovario obtuvieron una puntuación de 3, y los estilos en los que más del 75% de los tubos polínicos llegaron al ovario obtuvieron una puntuación de 4.
Adquisición de imágen. Se tomaron imágenes de los estilos seleccionados usando un microscopio Zeiss Axiophot con el juego de filtros 01, usando un Zeiss AxioCam ICc 5. Las imágenes se hicieron usando el paquete de software Zeiss Zen 2.3 (edición azul). Durante la adquisición, la configuración se ajustó para minimizar el fondo. Se tomaron imágenes de los estilos con el objetivo x5 y se guardaron como archivos TIFF con una resolución de 2464 x 2056 píxeles con una profundidad de 24 bits. Luego se ensamblaron hasta ocho imágenes separadas utilizando el ensamblador de imágenes Panavue. El contraste y el brillo de los estilos ensamblados se ajustaron para crear las Figs. 1y un 3c.
Extracción de ADN. Para el análisis KASP de las poblaciones de mapeo, se enviaron muestras de hojas a VHLGenetics (Wageningen, Países Bajos) para la extracción de ADN utilizando kits sbeadex™ (LGC Genomics GmbH, Berlín, Alemania) de acuerdo con el protocolo proporcionado por el fabricante.
análisis KASP. VHLGenetics (Wageningen, Países Bajos) realizó un análisis de PCR específico de alelo competitivo (KASP™) utilizando ensayos KASP diseñados para ser específicos para los SNP que se segregan en nuestro material. Los ensayos KASP se realizaron de acuerdo con el protocolo proporcionado por el fabricante (LGC Genomics GmbH, Berlín, Alemania). Los resultados de los ensayos KASP se visualizaron mediante SNPviewer (disponible en lgcgroup.com/products/genotyping-software/snpviewer) para confirmar la segregación y la llamada de genotipo correctas.
Análisis de vinculación. Los haplotipos de progenitores femeninos autocompatibles se reconstruyeron a partir de los datos del genotipo mediante el análisis de las tasas de recombinación entre diferentes SNP. Estos datos se utilizaron para convertir las llamadas SNP en un formato "axb", en el que el haplotipo "a" está vinculado al alelo autocompatible de Sli, mientras que el haplotipo "b" está vinculado a un alelo autoincompatible de Sli. Los mapas de vínculos se crearon utilizando Joinmap 4.126 con población tipo DH y configuración por defecto.
Mapeo de QTL. Los datos de fenotipo se convirtieron en un rasgo numérico asignando 1 a cada genotipo autocompatible, 0 a cada genotipo autoincompatible y * a los genotipos para los que no se pudo determinar la compatibilidad. El mapeo de QTL se realizó utilizando el mapeo de intervalos en MapQTL27. Los resultados de MapQTL se usaron para generar gráficos QTL con Mapchart 2.328.
Análisis bioinformático. Para identificar los modelos genéticos correctos en el intervalo inicial de 27.37 kb, investigamos dos anotaciones genéticas separadas para el genoma de referencia DM4.04, la anotación PGSC y la anotación ITAG. Véase también Hirsch et al.29. Para confirmar la corrección de las anotaciones, realizamos búsquedas BLASTp con las secuencias de proteínas predichas de ambas anotaciones. Al comparar los mejores resultados en la búsqueda BLASTp con nuestra consulta, determinamos si todos los exones y dominios anotados en la secuencia de proteínas predicha estaban respaldados por proteínas similares en papa y otras especies de plantas. Además, las bibliotecas de RNA-seq disponibles públicamente en SPUD DB (disponible en solanaceae.plantbiology.msu.edu/cgi-bin/gbrowse/potato/) y el visor de datos del genoma NCBI (disponible en ncbi.nlm.nih.gov/genoma/gdv /navegador/) se utilizaron para determinar si los exones putativos tenían evidencia de expresión. Juntos, estos dos enfoques nos permitieron validar las estructuras intrón-exón de los modelos de genes en ambas anotaciones, lo que resultó en una elección informada de una o más isoformas de modelos de genes para representar el gen en cuestión. Sobre la base de estos enfoques, el gen candidato PGSC0003DMG400016862 se reconoció como probablemente expresado de forma parcial e insignificante y se descartó de análisis posteriores. El modelo de gen Sotub12g029970 se consideró correcto, mientras que su homólogo de PGSC, PGSC0003DMG400016860, probablemente esté truncado. Debido a que está ubicado en gran medida fuera del intervalo designado, y no se pudieron identificar sustituciones de aminoácidos relevantes entre las plantas SC y SI, este gen se descartó de análisis posteriores.
Análisis de variación. Para identificar mutaciones en el intervalo de 27.37 kb que son específicas para genotipos autocompatibles, se determinaron todos los SNP de alta confianza (Datos complementarios 3) que eran (1) homocigotos en DS, 17SC100-18 y 17SC100-2 (porque los tres son homocigotos para el alelo SC de Sli (Sli/Sli')'), (2) homocigotos diferentes en D16 (porque D16 es homocigoto para el alelo SI de Sli (sli/sli)), y (3) heterocigotos tanto en 16HP1-66 como en 17SC25-8 (porque ambos son heterocigotos para el alelo SC Sli (Sli /sli)). La secuencia alélica se obtuvo por ensamblaje de novo usando SPAdes versión 3.11.130 de 150 nt de datos de Illumina de extremo emparejado de las plantas enumeradas anteriormente (de aproximadamente 25-30X de profundidad de secuenciación). Los contigs resultantes se alinearon con la referencia de DM (usando minimap2 versión 2.1) y se filtraron para aquellos que se alineaban de manera confiable con los 27 kb. A partir de estos contigs alineados, la variación relativa a DM4.03 se cuantificó de manera directa (usando las subrutinas compilación y llamada de bcftools, versión 1.9) y se enumeró en el formato de llamada variante (VCF).
Análisis de cambio de aminoácidos. De esta lista de mutaciones específicas de SC, todos los SNP no sinónimos se identificaron mediante la superposición con los exones de codificación designados. Se enumeraron los cambios de aminoácidos relativos a la secuencia de DM o SI. Los cambios de aminoácidos únicos se identificaron realizando búsquedas BLASTp usando la secuencia de la proteína y realizando múltiples alineaciones de secuencias usando los 100 mejores resultados de BLASTp.
Variación en las regiones promotora y terminadora. La región promotora se eligió para ser la secuencia cadena arriba del codón de inicio hasta la secuencia codificante del gen cadena arriba con un máximo de 1500 nt. Se encontró una variación dramática en las regiones promotoras dentro del intervalo de 27.37 kb, de las cuales las más llamativas fueron varias deleciones e inserciones más grandes de decenas a cientos de nucleótidos de longitud. Se obtuvo toda la variación en el intervalo Sli, en relación con DM, incluida la del promotor/región aguas arriba así como la región terminadora/aguas abajo.
Adquisición y germinación de polen. El polen de los genotipos enumerados en la Fig. 2a se obtuvo vibrando flores abiertas con un cepillo de dientes electrónico y recolectando el polen en tubos Eppendorf de 1.5 ml. Después de la adquisición, el polen se secó almacenando los tubos Eppendorf abiertos con polen en una caja hermética que contenía gel de sílice durante 24 horas a temperatura ambiente. Posteriormente, el polen se almacenó a -20 °C hasta su uso posterior.
El polen se germinó suspendiendo 2.5 mg de polen seco en 5 ml de medio líquido (9% (p/v) de sacarosa, 50 mg/l de ácido bórico, 73.5 mg/l de CaCly2H2O, 118 mg/l Ca (NO3) 24H2O, 123 mg/l MgSO.4VH2O) en cajas Petri de 3.5 cm de diámetro selladas con parafilm. El polen se dejó germinar en las cajas de Petri durante 24 h en oscuridad en una incubadora con agitación a temperatura ambiente y agitación a 125 RPM. A continuación, el medio líquido que contenía el polen germinado se pipeteó con cuidado en tubos Eppendorf de 2 ml usando puntas de pipeta que se modificaron para aumentar el tamaño de la abertura para no dañar los tubos polínicos. A continuación, los tubos Eppendorf se centrifugaron a 600 xg durante 1 min y el medio se retiró con cuidado mediante pipeteo. El sedimento y parte del medio restante se congelaron inmediatamente en nitrógeno líquido, se agregaron dos perlas de acero inoxidable (2 mm de diámetro) y las muestras se trituraron utilizando un TissueLyser II (Qiagen GmbH, Hilden, Alemania) a 20 Hz durante 1 min.
Extracción y secuenciación de ARN. Se añadió tampón RLT (Qiagen GmbH) a las muestras de polen molido mientras se aseguraba de que las muestras permanecieran congeladas. Luego se realizó la extracción de ARN utilizando el mini kit RNeasy de acuerdo con el protocolo del fabricante (Qiagen GmbH, Hilden, Alemania). Las bibliotecas de ADNc de tamaño de inserción de 250-300 pb se secuenciaron como lecturas de extremos emparejados de 150 nt, lo que produjo 30-42 millones de pares de lectura por muestra (Novogene, Cambridge, Reino Unido).
Otros conjuntos de datos de RNA-seq. Para crear una descripción general de los niveles de expresión (específicos de tejido), todos los conjuntos de datos de RNA-seq secuenciados por pares etiquetados como ORGANISMO “Solanum tuberosum” se descargaron del dominio público (NCBI-SRA, fecha 2018/17/13), con un total de 441 conjuntos de datos fastq emparejados. De estos 441 conjuntos de datos públicos, 3 se generaron a partir de tejido de estilo (SRR7402817-SRR7402819) y todos los demás a partir de varios tejidos sin polen, etapas de desarrollo y accesiones de plantas.
Conjunto de referencia de Solyntus. Para los análisis de expresión, el borrador recientemente adquirido de la línea de referencia homocigótica Solyntus (versión 1.0, descargable en www.plantbreeding.wur.nl/Solyntus/) se utilizó como genoma de referencia. Solyntus es una variedad esencialmente homocigota generada como parte del programa de mejoramiento de Solynta21. Los intervalos de mapeo en este estudio se infirieron del ensamblaje del genoma de DM v4.0331 al ensamblaje del genoma de Solyntus 1.0 mediante búsquedas de similitud básicas (usando BLASTn y bedtools) para ubicarse en (coordenadas del ensamblaje del genoma de Solyntus 1.0) 53532708-53954293 (Intervalo I, 421.6kb < —628.9kb), 53683239-53867377 (Intervalo II, 184.1 kb< — 168.7 kb), 53731620-53763003 (Intervalo III, 31.4 kb < —27.4 kb) y 53753977-53763003 (Intervalo IV, 9.0 kb < —12.6 kb), respectivamente. Entre paréntesis se encuentran el número de intervalo de mapeo consecutivo [coordenadas de Solyntus 1.0], el tamaño en Solyntus-1.0 y el tamaño en DM-4.03/4.04, respectivamente. Todos los intervalos están ubicados en el cromosoma ST4.03ch12_RaGOO (que es el cromosoma 12) y no contienen un solo espacio en el ensamblaje de Solyntus 1.0. La variación del tamaño del intervalo está causada por una multitud de lagunas (N) en la secuencia de DM correspondiente y una gran variación entre ambos genomas. Intervalos correspondientes en el genoma de DM (DM-4.03/4.04): Intervalo I: chr12:58601503-59230363, Intervalo II: chr12:58962004-59130723; Intervalo III: 59016142-59043512; Intervalo IV: chr12:59030880-59043512.
La anotación de genes en Solyntus 1.0 se dedujo de tres catálogos de genes distintos (potato DM4.03, ITAG4.0 Tomato Genome Annotation Release del 6 de septiembre de 201932y Pepper-v. 1.5533), que se mapearon en el ensamblado de Solytus mediante GeMoMa (v1.6.1). Esto se hizo para compensar las imperfecciones en los catálogos de genes individuales y maximizar nuestro conocimiento de la existencia de posibles genes y/o loci expresados.
Mapeo de lectura de RNA-seq y cantidad de abundancia de transcripciónficatión. Los 5 SC, 3 SI y los 441 conjuntos de datos públicos de RNA-seq se asignaron al genoma de referencia de Solyntus utilizando hisat2 (versión 2.1.0). El catálogo de genes híbridos obtenido con GeMoMa se usó para la estimación de abundancia guiada por transcripción con StringTie (versión 2.1.1) con configuraciones -t -c 5 -f 0.05 -G y un archivo Solyntus1.0 gff concatenado con GeMoMa. Toda la expresión observada en un intervalo de 500 kb que rodea al SLI se evaluó locus como centro, en cuyo intervalo se ubican un total de 90 loci de genes (inferidos). Confirmamos la ausencia de cualquier expresión notable en muestras SC fuera de cualquiera de estos loci de genes. En el intervalo de 500 kb, indicamos el número subsiguientemente menor de genes candidatos cuando se cruzan con nuestros intervalos de mapeo I-IV como se definió anteriormente.
Confirmación de haplotipo-specifiexpresión c. De 90 loci expresados en el intervalo de 500 kb, solo 8 se expresaron por encima de un umbral seleccionado de 20 FPKM en todas las muestras SC/SI. Utilizamos estos sitios para medir las diferencias de nivel de expresión específicas de haplotipo (Sli o sli). El umbral de expresión seleccionado permitió una profundidad de lectura suficiente para eventualmente y de manera confiable la expresión en (como máximo) 2 haplotipos. Junto con el propio locus PSC (que carece de expresión en plantas SI), estos 8 + 1 loci fueron haplotipados en cada una de las 8 muestras (SAMtools fase versión 1.7, configuración predeterminada). Los archivos fastq haplotipados (emparejados) resultantes se ensamblaron de novo utilizando SPAdes (versión 3.11.1). Los contigs resultantes se filtraron en busca de una gran abundancia y se supusieron ARNm de longitud completa, correspondientes a la isoforma expresada principal (haplotipada). En algunos casos, esto eliminó isoformas empalmadas alternativamente, ninguna de las cuales fue respaldada por amplias lecturas como de importancia biológica obvia. La variación en estas secuencias de ARNm haplotipadas se usó para (des)confirmar si uno o ambos haplotipos se expresaban en cada uno de los loci/muestras correspondientes.
Diseño de SLI construcción de expresión. Usamos la secuencia de la planta donante de Sli DS para diseñar el casete de expresión de Sli. Para permitir la expresión nativa de psc, construimos una secuencia de ácido nucleico que comprende el promotor nativo (1563 pb aguas arriba del codón de inicio), los tres exones y el terminador nativo (740 pb aguas abajo del codón de parada). Por lo tanto, ambos intrones fueron eliminados del PSC gen de la planta donante DS. Esta secuencia fue sintetizada y clonada en pBINPLUS por Genscript (Genscript Biotech, Leiden, Países Bajos). Nos referimos al vector que contiene el SLI insertar como pBINPLUS-Sli.
Construcción de CRISPR–vector cas9. Diseñamos cuatro ARNg basados en la secuencia de PGSC0003DMG400016861 en DM4.03 en lugares en los que no había variación entre los alelos SC y SI. Para la selección de guías adecuadas y construcción del vector se utilizó el método descrito por Santillán Martínez et al.34. En resumen, se seleccionaron cuatro sgRNA de acuerdo con las pautas descritas por Liang et al.35. Se utilizó la herramienta de predicción de objetivos CC-Top CRISPR/Cas9 para generar una lista de sgRNA36, el plegado se evaluó utilizando el servidor web Mfold37, y la actividad de los sgRNA se predijo utilizando el marcador de sgRNA38. Se seleccionaron y usaron las siguientes guías para construir el vector pAGM:CRISPRASli: exon5.1T01 (ATTTCATCCGCGATCTCTCGGGG), exon5.1T04 (GATTTCA TCCGCGATCTCTCGGG), exon5.1T06 (TATTTCCTATTGCTACCAGAAGG) y exon5.1T07 (TGATTTCATCCGCGATCTCTCGG). A continuación, la construcción CRISPR se sintetizó utilizando plásmidos obtenidos de Addgene: pICH86966 (plantilla para amplificación); pICSL01009 (plásmido de nivel 0); pICH47751, pICH47761, pICH47772, pICH47781 y pICH47732 (plásmidos de nivel 1); pICH41822 (enlazador para cuatro guías); y pAGM4723 (vector binario de nivel 2). El plásmido fue clonado usando E. coli DH5a y el plásmido purificado se enviaron para la secuenciación utilizando los cebadores PDS5843 (TTTGTGATGCTCGTCAGGGG), PDS8535 (CCCGAGAATTATGCAGCATT TT) PDS8536 (TCATCAGTCAATTACGGGGCT) y AL717 (GCTTGGCATC AGACAAACCGG) para confirmar la presencia y la orientación correcta de todos los componentes (NPTII, Cas9 y sgRNA).
Transformación de pBINPLUS-Sli y pAGMzCRISPRADeslice el vector en Agrobacterium tumefaciens. Transformamos pBINPLUS-Sli en A. tumefaciens colar AGL0 y pAGM:CRISPRASli en A. tumefaciens cepas AGL0 y AGL1 utilizando un protocolo de electroporación. Tomamos 40 pl de células AGL0 competentes y añadimos 110 pl de agua milliQ helada. Pipeteamos 50 µl de esta mezcla en tubos Eppendorf preenfriados en hielo y añadimos 1 µl del plásmido. Dejamos las células en hielo durante 15 minutos y transferimos las células a cubetas de electroporación preenfriadas. Electroporamos las mezclas con un Micropulser™ (Bio-Rad Laboratories, Vee-nendaal, Países Bajos) utilizando el programa Ec1 (1.8 kV, cubeta de 0.1 cm). Añadimos 1 ml de LB e incubamos las células durante 3 h en un agitador a 28 °C y 200 RPM. Posteriormente, inoculamos placas de agar LB que contenían Rifampicina (100 pg/ml) y Kanamicina (50 pg/ml) con el cultivo de transformación. Se confirmó que todas las colonias seleccionadas contenían el vector correcto.
Transformación de genotipos de papa. Transformamos los genotipos B666 y B667 con pBINPLUS-Sli y los genotipos B663 y B665 con el vector pAGM:CRISPRASli utilizando el método de explante de tallo descrito por Visser39. Brevemente, se obtuvieron explantes de entrenudos a partir de genotipos cultivados in vitro y se colocaron en placas de Petri que contenían medio R3B con 2 ml de medio PACM. Al día siguiente, 50 ml de 48 h Agrobacterium los cultivos se centrifugaron y se resuspendieron en 75 ml de LB. A continuación, los explantes de entrenudos se sumergieron en el Agrobacterium suspensión durante 5 min, se secó en el filtro y se volvió a colocar en las placas de Petri que contenían medio R3B. Después de 48 h de incubación, los explantes se transfirieron a cajas de Petri que contenían MS20 con antibióticos y se colocaron en una cámara de crecimiento para permitir la regeneración de brotes. Después de la regeneración, los brotes se cultivaron en medio MS20 que contenía cefotaxima (200 pg/ml), vancomicina (200 pg/ml) y kanamicina (100 pg/ml). Cuando los brotes alcanzaron la longitud suficiente, se hicieron esquejes y se cultivaron en MS20 sin antibióticos. Después de al menos dos semanas de cultivo en MS20 sin antibióticos, las plantas se plantaron en el invernadero.
Análisis de ploidía. La ploidía de las plantas transgénicas, así como de los controles no transformados, se determinó mediante citometría de flujo por Plant Cytometry Services (Didam, Países Bajos). Todos los regenerantes tetraploides fueron descartados.
Análisis de PÁGINA de CRISPR–Mutaciones inducidas por Cas9. La extracción de ADN, PCR y análisis PAGE fueron realizados por Limgroup (Horst, Países Bajos). Se usaron los siguientes cebadores para amplificar la región dirigida por CRISPR-Cas9: Cebador directo: CTATTTCCTATTGCTACCAG, cebador inverso: AAACTTTACCCAAAT AACGTC. El marcaje de los productos de la PCR se logró agregando un cebador inverso con una cola M13 (secuencia del cebador: TGTAAAACGACGGCCAGTAAACTTTAC CCAAATAACGTC) y 700 cebadores M800 marcados con IRDye u 13 con IRDye en la mezcla de PCR. Los productos de PCR resultantes se analizaron en PAGE utilizando un sistema Li-cor. Los productos de PCR de la mayoría de las líneas sin mutaciones inducidas por CRISPR-Cas9 se eliminaron de las imágenes del gel para producir la Fig. 3d (excisiones indicadas por líneas discontinuas).
Análisis filogenético de la inserción de 533 pb.. La secuencia de la inserción de 533 pb se analizó mediante BLASTn en el sitio web del NCBI. Los mejores 30 éxitos, incluida la inserción presente en SLI en Solyntus, luego se descargaron y alinearon en MegAlign Pro 17 (DNASTAR) usando MUSCLE con la configuración predeterminada. Los árboles se generaron utilizando el algoritmo de unión de vecinos con la configuración predeterminada.
Resumen de informes. Más información sobre el diseño de la investigación está disponible en el Resumen de informes de investigación de Nature vinculado a este artículo.
Disponibilidad de datos
La secuencia del genoma de Solyntus y las lecturas de la secuencia sin procesar están disponibles en NCBI bajo acceso PRJNA631911. Los archivos de ensamblaje y anotación del genoma de Solyntus están disponibles en WUR [https://www.plantbreeding.wur.nl/Solyntus/]. Los datos de secuenciación de ARN del polen germinado están disponibles en el archivo de lectura breve del NCBI bajo acceso PRJNA713577. Otros datos están disponibles en el archivo de datos de origen o estarán disponibles a pedido. Los datos de origen se proporcionan con este documento.
Recibido: 22 de enero de 2021; Aceptado: 8 de junio de 2021;
Publicado en línea: 06 Julio 2021
Referencias
- 1. Duvick, DN La contribución del mejoramiento a los avances en el rendimiento del maíz (Zea mays l.). Adv. Agrón. 86, 83-145 (2005).
- 2. Lindhout, P. et al. Hacia el mejoramiento de papas de siembra híbridas F1. Patata Res. 54, 301-312 (2011).
- 3. Jansky, SH et al. Reinventando la papa como un cultivo basado en línea endogámica diploide. Ciencia de cultivos 56, 1412-1422 (2016).
- 4. Vosotros, M. et al. Generación de patata diploide autocompatible por knockout de S-RNase. Nat. Plantas 4, 651-654 (2018).
- 5. Enciso-Rodríguez, F. et al. Superando la autoincompatibilidad en papa diploide usando CRISPR-cas9. Frente. Plant Sci. 10, 1-12 (2019).
- 6. Su, Y. et al. Introgresión de genes de resistencia contra Phytophthora infestans en papa diploide. Soy. J. Patata Res. 97, 33-42 (2020).
- 7. Dzidzienyo, DK, Bryan, GJ, Wilde, G. & Robbins, TP Diversidad alélica de alelos S-RNasa en especies diploides de papa. teor. aplicación Gineta. 129, 1985-2001 (2016).
- 8. McClure, B., Cruz-Garcia, F. & Romero, C. Compatibilidad e incompatibilidad en sistemas basados en S-RNasa. Ana. Bot. 108, 647-658 (2011).
- 9. Kubo, K. et al. Sistema colaborativo de no auto reconocimiento en autoincompatibilidad basada en S-RNase. Ciencia: 330, 796-799 (2010).
- 10. De Jong, H. & Rowe, PR Consanguinidad en papas diploides cultivadas. Patata Res. 14, 74-83 (1971).
- 11. Hermsen, JGT & Olsder, J. Genética de la autocompatibilidad en dihaploides de Solanum tuberosum L. 1. Comportamiento reproductivo de dos dihaploides autocompatibles. eufítica 25, 597-607 (1976).
- 12. Hosaka, K. & Hanneman, RE Jr. Genética de la autocompatibilidad en una especie de patata diploide silvestre autoincompatible Solanum chacoense. 1. Detección de un gen inhibidor del locus S (Sli). eufítica 99, 191-197 (1998).
- 13. Hosaka, K. & Hanneman, RE Jr. Genética de la autocompatibilidad en una especie de patata diploide silvestre autoincompatible Solanum chacoense. 2. Localización de un gen inhibidor del locus S (Sli) en el genoma de la patata utilizando marcadores de ADN. eufítica 103, 265-271 (1998).
- 14. Birhman, RK & Hosaka, K. Producción de progenies puras de papas diploides usando un gen inhibidor del locus S (Sli) y su caracterización. Genoma 502, 495-502 (2000).
- 15. Chen, J., Hu, Q., Zhang, Y., Lu, C. y Kuang, H. P-MITE: una base de datos para elementos transponibles de repetición invertida en miniatura de plantas. Nucleic Acids Res. 42, 1176-1181 (2014).
- 16. Stefanowicz, K., Lannoo, N. & Van Damme, EJM Plant F-box proteins— juzga entre la vida y la muerte. crítico Rev. Plant Sci. 34, 523-552 (2015).
- 17. Liu, B., Morse, D. y Cappadocia, M. La glicosilación de S-RNasas puede influir en los umbrales de rechazo de polen en Solanum chacoense. Exp. J. Bot. 59, 545-552 (2008).
- 18. Laimbeer, FPE La genómica de la papa de tres maneras: cuantificación de la endorreduplicación en los tubérculos, un recorrido por el terreno del transposón y elucidación de la regulación del color de las flores. http://hdl.handle.net/10919/84480 (2018).
- 19. Clot, CR et al. El origen y la ocurrencia generalizada de autocompatibilidad basada en Sli en papa. teor. aplicación Gineta. https://doi.org/10.1007/s00122-020-03627-8 (2020).
- 20. Endelman, J., Jansky, SH, Butler, N. & Christensen, G. Evidencia genética de un alelo letal recesivo en el informe anual del cromosoma 12 de la papa de la Asociación de la Papa de América. Soy. J. Patata Res. 96, 331 (2019).
- 21. van Lieshout, N. et al. Solyntus, el nuevo genoma de referencia altamente contiguo para la papa (Solanum tuberosum). G3 Genes Genomas Genet. 10, 3489-3495 (2020).
- 22. Leisner, CP et al. Secuencia del genoma de M6, un clon endogámico diploide de la especie de papa productora de tubérculos con alto contenido de glicoalcaloides Solanum chacoense, revela heterocigosidad residual. Planta J. 1967, 562-570 (2018).
- 23. Peterson, BA et al. Autofertilidad en una población de papa diploide cultivada examinada con la matriz de polimorfismo de un solo nucleótido de papa infinium 8303. genoma vegetal https://doi.org/10.3835/plantgenome2016.01.0003 (2016).
- 24. Lian, Q. et al. Adquisición de mutaciones deletéreas durante la poliploidización de la papa. J.Integr. Biol. vegetal 61, 7-11 (2019).
- 25. van Lieshout, N. et al. Solyntus, el nuevo genoma de referencia altamente contiguo para la patata (Solanum tuberosum). G3 Genes Genomas Genet. 631911, g3.401550.2020 (2020).
- 26. Van Ooijen, JW UnirseMapa®4, Software para el Cálculo de Mapas de Ligamiento Genético en Poblaciones Experimentales vol. 33 (Kyazma BV, 2006).
- 27. van Ooijen, JW Precisión del mapeo de loci de rasgos cuantitativos en especies autógamas. teor. Aplicación Genet. 84, 803-811 (1992).
- 28. Voorrips, RE Mapchart: software para la presentación gráfica de mapas de ligamiento y QTL. J. Hered. 93, 77-78 (2002).
- 29. Hirsch, CD et al. Spud DB: un recurso para extraer secuencias, genotipos y fenotipos para acelerar el mejoramiento genético de la papa. genoma vegetal. 7, https://doi.org/ 10.3835/plantgenoma2013.12.0042 (2014).
- 30. Bankevich, A. et al. SPAdes: un nuevo algoritmo de ensamblaje del genoma y sus aplicaciones a la secuenciación unicelular. J. Cómputo. Biol. 19, 455-477 (2012).
- 31. Sharma, SK et al. Construcción de pseudomoléculas de escala cromosómica de referencia para papa: integración del genoma de papa con mapas genéticos y físicos. G3 Genes Genomas Genet. 3, 2031-2047 (2013).
- 32. Fernández-Pozo, N. et al. The Sol Genomics Network (SGN): del genotipo al fenotipo y al mejoramiento. Nucleic Acids Res. 43, D1036-D1041 (2015).
- 33. Kim, S. et al. La secuencia del genoma del pimiento picante proporciona información sobre la evolución de la acritud en Pimiento especies. Nat.. Genet. 46, 270-278 (2014).
- 34. Santillán Martínez, MI et al. Mutagénesis dirigida por CRISPR/Cas9 del gen de susceptibilidad del tomate PMR4 para la resistencia contra el mildiú polvoroso. BMC Planta Biol. 20, 1-13 (2020).
- 35. Liang, G., Zhang, H., Lou, D. y Yu, D. Selección de sgRNA altamente eficientes para la edición del genoma de plantas basado en CRISPR/Cas9. Sci. Reps. 6, 1-8 (2016).
- 36. Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J. & Mateo, JL CCTop: una herramienta de predicción de objetivos CRISPR/Cas9 intuitiva, flexible y confiable. PLoS ONE 10, 1-11 (2015).
- 37. Zuker, M. Servidor web Mfold para el plegamiento de ácidos nucleicos y la predicción de hibridación. Nucleic Acids Res. 31, 3406-3415 (2003).
- 38. Chari, R., Yeo, NC, Chavez, A. & Church, GM SgRNA Scorer 2.0: un modelo independiente de especie para predecir la actividad CRISPR/Cas9. Sintetizador ACS. Biol. 6, 902-904 (2017).
- 39. Visser, RGF en Manual de cultivo de tejidos vegetales 301-309 (Springer, 1991).
Conflicto de intereses
Los autores declaran no tener conflictos de intereses.
Información Adicional
Información suplementaria La versión en línea contiene material complementario disponible en https://doi.org/10.1038/s41467-021-24267-6.
Correspondencia y las solicitudes de materiales deben dirigirse a CWBB
Información de revisión por pares Nature Communications agradece a Roger Chetelat y a los otros revisores anónimos por su contribución a la revisión por pares de este trabajo. Los informes de revisión por pares están disponibles.
Reimpresiones e información de permisos está disponible en http://www.nature.com/reprints
Publisher 's nota Springer Nature permanece neutral con respecto a los reclamos jurisdiccionales en mapas publicados y afiliaciones institucionales.
Agradecimientos
Agradecemos la ayuda de Veronica Tammy Soputro en la transformación de dos genotipos de papa con pAGM:CRISPRASli, la ayuda de los estudiantes de licenciatura y maestría Gilo Pleunis, Iris Smits, Niki Vorgia, Hidde Knuiman, Maurice Geurts, Anja van Heteren y Torsten van der Schriek por su ayuda con el fenotipado de las poblaciones de mapeo y a Tess Lucas por generar muestras de ARN. También agradecemos a los empleados de los invernaderos Unifarm de la Universidad de Wageningen y Solynta por su ayuda con el mantenimiento de las plantas y la extracción de semillas. Este proyecto ha recibido apoyo financiero de la Organización Holandesa de Investigación Científica (ID de subvención: NWA.17.023).
Contribuciones de autor
E.-JE diseñó y ejecutó los experimentos y escribió el manuscrito. AvdB diseñó y ejecutó los enfoques WGS, KASP y bioinformático. SvH, RGFV, CWBB, MEdV y PL ayudaron a diseñar el enfoque para los estudios de mapeo genético y la caracterización funcional de SLI. RGFV, CWBB y PL revisaron y comentaron el manuscrito.
yo(s) q) Acceso Abierto Este artículo está licenciado bajo una licencia internacional Creative Commons Attribution 4.0, que permite el uso, intercambio, adaptación, distribución y reproducción en cualquier medio o formato, siempre que otorgue el crédito apropiado al autor o autores originales y a la fuente, proporcione un enlace a la licencia Creative Commons e indique si se realizaron cambios. Las imágenes u otro material de terceros en este artículo se incluyen en la licencia Creative Commons del artículo, a menos que se indique lo contrario en una línea de crédito al material. Si el material no está incluido en la licencia Creative Commons del artículo y su uso previsto no está permitido por la regulación legal o excede el uso permitido, deberá obtener el permiso directamente del titular de los derechos de autor. Para ver una copia de esta licencia, visite http://creativecommons.org/ licencias/por/4.0/.




